Stargardt Disease – May, 2022

History:
A 32-year-old woman was referred by an optometrist for macular changes in both eyes. She noted intermittent mild blurriness she attributed to dryness but was otherwise asymptomatic. Her past medical history was significant for migraines. She denied any family history of any ocular diseases. Review of systems was non-contributory.
Exam:
Her visual acuity was 20/20 and 20/20 in the right and left eye, respectively. There was no RAPD. Intraocular pressures were 21 and 18 in the right and left eyes, respectively. Anterior segment exam demonstrated clear crystalline lenses in both eyes. Dilated fundus examination demonstrated many pisciform (fish-shaped) lesions centered within the posterior pole. See imaging below.
Figure 1: Fundus photographs of the right eye and left eyes demonstrated many pisciform lesions centered within the posterior pole.
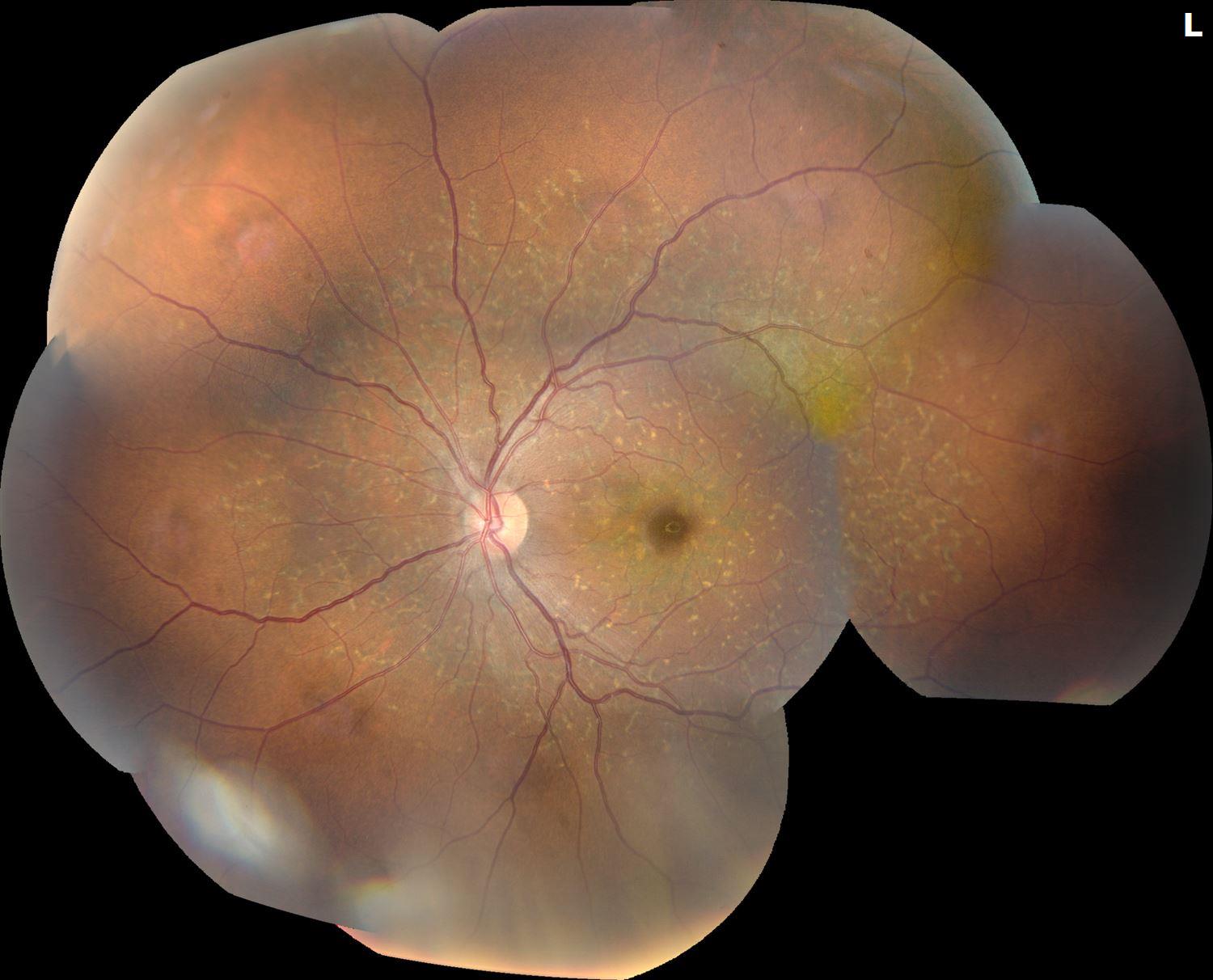
Figure 2: OCTscans of the right and left eyes demonstrated the presence of the accumulation of hyperreflective material within the RPE as well as some disruption of the photoreceptor layer.
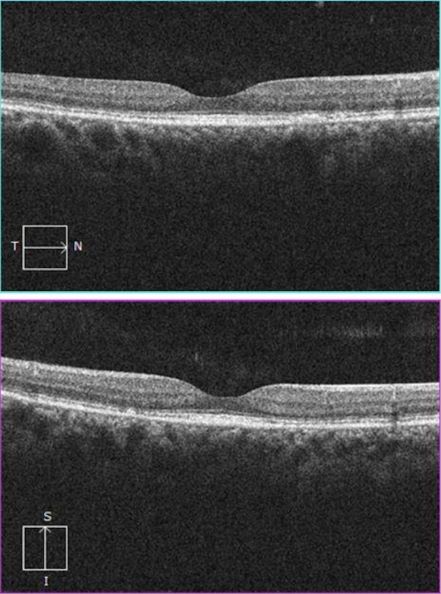
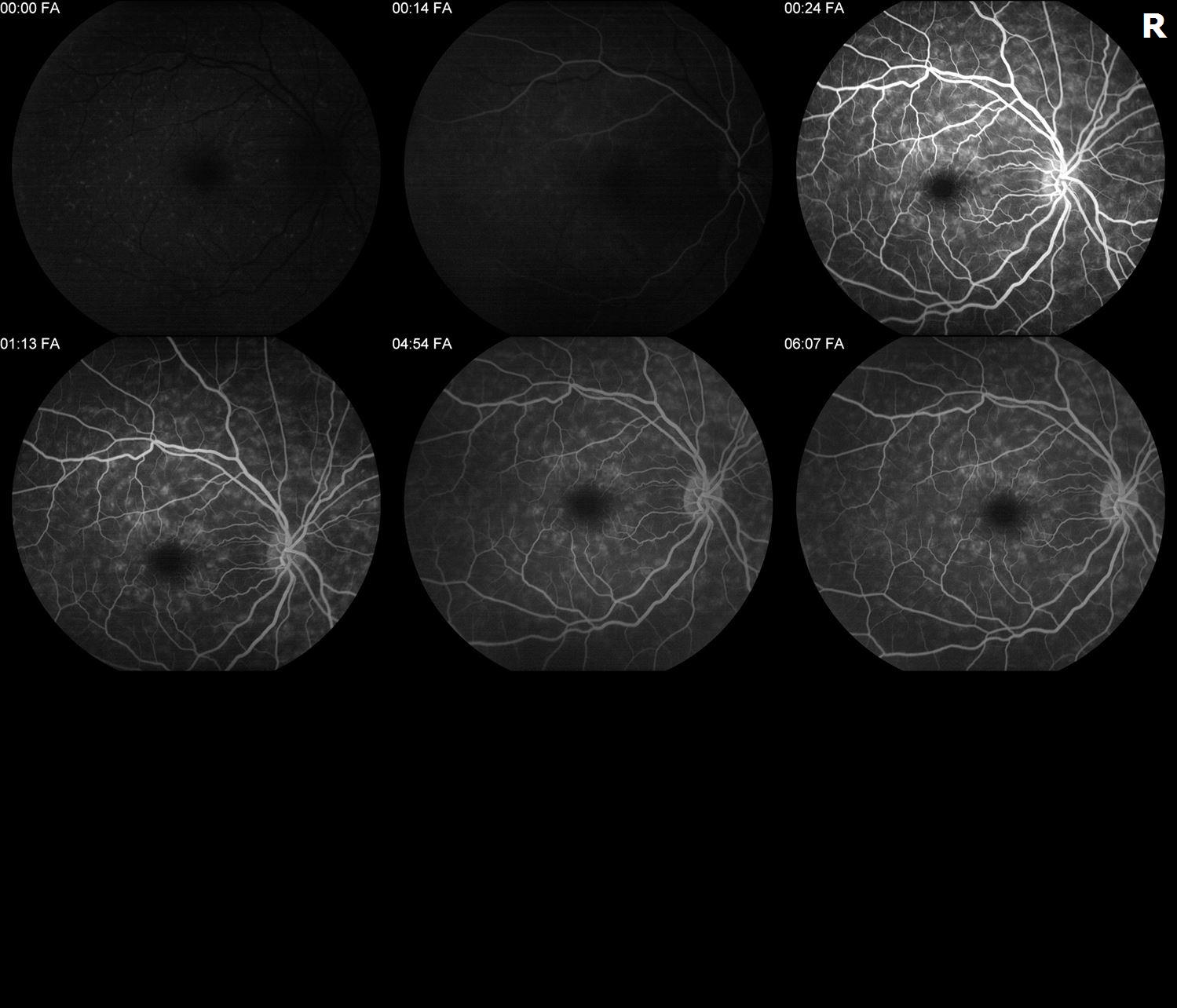
Figure 3: Intravenous fluorescein angiography showed a darkened choroid and hyperfluorescence of the pisciform lesions.
Differential Diagnosis:
- Age-related macular degeneration
- Stargardt disease
- Pattern dystrophy
- Central areolar choroidal dystrophy
- Cone dystrophy and Cone rod dystrophy
- Best disease
Discussion:
Stargardt Disease
Stargardt disease is the most common childhood, recessively inherited macular dystrophy and was described by Karl Stargardt in 1909. Stargardt disease is caused by mutations to the ABCA4 gene and causes the accumulation of visual cell cycle by-products to accumulate in the RPE and cause secondary photoreceptor dysfunction and eventual cell death. The condition affects 1 per 10,000 individuals and usually manifests in childhood or adolescence but can occur later such as in our case. Usually, a later age of onset will result in a milder disease course. Some cases may be asymptomatic however most present with central vision loss between childhood and adolescence. Typically, the vision at presentation ranges from 20/20 to 20/400 which may explain our patients relatively late presentation with her excellent vision. In rare cases, a small minority of patients can progress to hand motion vision.
Stargardt disease typically affects the macula with variable expansion into the posterior pole. Early in the course of the disease, the exam may be normal. Later, the typical fundus findings arise with pigmentary mottling, macular atrophy, and pisciform flecks. OCT will demonstrate accumulation of hyperreflective material within the RPE as well as photoreceptor disruption and loss. Fluorescein angiography may demonstrate a “dark-choroid” sign in up to 60% of patients since the lipofuscin will block the choroidal fluorescence. Additionally, the flecked lesions will appear hyperfluorescent. Genetic testing confirms the diagnosis.
The mainstay of current management involves UV protective sunglasses as well as low vision aids. Studies in mice suggest patients with this condition may have a slower progression of disease with less light exposure, however this has not been studied in human trials. This condition remains an active target for gene therapy trials.
The patient underwent genetic testing which confirmed their presumed diagnosis. UV protective sunglasses were recommended, and the patient will follow up for annual eye exams. Their genetic testing will serve important for family planning or to identify them as a possible candidate for a future gene therapy.
If you are looking to schedule your first consultation, please contact us today by clicking HERE and find the location that is nearest you!
References:
- Fujinami K, Lois N, Davidson AE, et al. A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am J Ophthalmol 2013;155:1075–88.
- Strauss RW, Ho A, Munoz B, et al. The natural history of the progression of atrophy secondary to Stargardt disease (ProgStar) studies: design and baseline characteristics: ProgStar Report No. 1. Ophthalmology 2016;123:817–28.
- Haji Abdollahi S, Hirose T. Stargardt-Fundus flavimaculatus: recent advancements and treatment. Semin Ophthalmol 2013;28:372–6.
- Fishman GA, Stone EM, Grover S, et al. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol 1999;117:504–10.